在合成化学与生命科学中,获得对映体纯的分子对于催化剂、配体及药物分子的性能至关重要。然而,许多利用直接电解的有机反应虽然具备原子经济和操作简便的优势68配资,却因缺乏手性诱导而难以摆脱“非对映选择性”的瓶颈。尤其是当高活性 自由基或离子‑自由基中间体在电极‑电解质界面瞬时生成时,传统的手性催化剂往往来不及干预,以致反应物沿着本征的消旋轨道前进。如何在不依赖额外手性金属或有机催化剂的情况下,于直接电解体系中实现高对映选择性,长期被视为电化学合成领域的一大挑战.
鉴于此,康奈尔大学林松教授提出以“亚化学计量量手性磷酸盐支撑电解质”引导动态动力学拆分(dynamic kinetic resolution, DKR),在恒流直接电解条件下将外消旋三价膦氧化为高对映富集的膦氧化物。策略核心在于:电阳极一次电子氧化快速生成构型可逆的磷鎓自由基阳离子,随后其在电极附近高浓度手性磷酸根包围的微环境中发生速率决定的亲核加成,最终得到对映体比(e.r.)最高可达 99:1 的产物。该手性支撑电解质既充当导电载体,又通过离子对作用实现对映诱导,与流动电解技术结合时用量可进一步降低,展现了“绿色、经济且可回收”的潜力。相关研究成果以题为“Dynamic kinetic resolution of phosphines with chiral supporting electrolytes”发表在最新一期《nature》上。

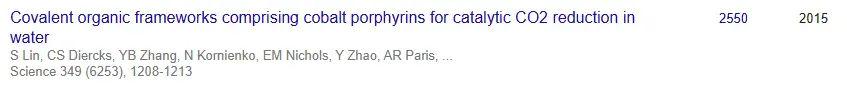
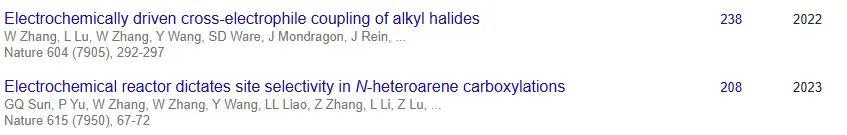
值得一提的是,这也是林松教授发表的第8篇NS。

林松,2008年本科毕业于北京大学化学系,2013年博士毕业于美国哈佛大学化学专业,2016年完成了在加州大学伯克利分校的博士后研究员工作后,加盟康奈尔大学至今,他目前为康奈尔大学教授。凭借其突出的科研贡献,他自2019年起已获得了多个国际奖,收获了众多荣誉。他曾获2019年美国斯隆研究奖(化学领域),2019年度《麻省理工科技评论》中国区“35 岁以下科技创新 35 人”;去年更是获得了含金量颇高的美国化学会颁发的亚瑟科普学者奖。近年来,林松教授利用有机化学和电化学的方法开发的催化剂,实现了高效的化学合成和材料转化。他用电化学取代传统制药路径的发现,当时被誉为“过去十年最有价值反应”,预估可给相关能源和制药公司带来相当可观的利益,同时还能减少化学合成污染和全球的碳排放,有利于缓解全球温室效应




【反应发展】
作者首先比较了三条可行途径:手性电极、手性溶剂与手性支撑电解质(图1)。手性溶剂仅适用于毫克级反应;手性电极在重复性与优化空间上受限;而手性支撑电解质既能独立调节阳离子/阴离子结构,又因电双层富集效应使手性离子在阳极面浓度提升约三倍,从而放大对映选择。模型体系中,三价膦被氧化为磷鎓自由基阳离子,其锥面翻转能垒仅 ≈5 kcal mol⁻¹,远低于中性膦的 30–40 kcal mol⁻¹,确保在亲核加成前完成快速外消旋;随后手性磷酸根与其形成紧密离子对,引导产生主产物的速率常数 kfast 显著高于副产物 kslow,奠定了DKR放大效应。
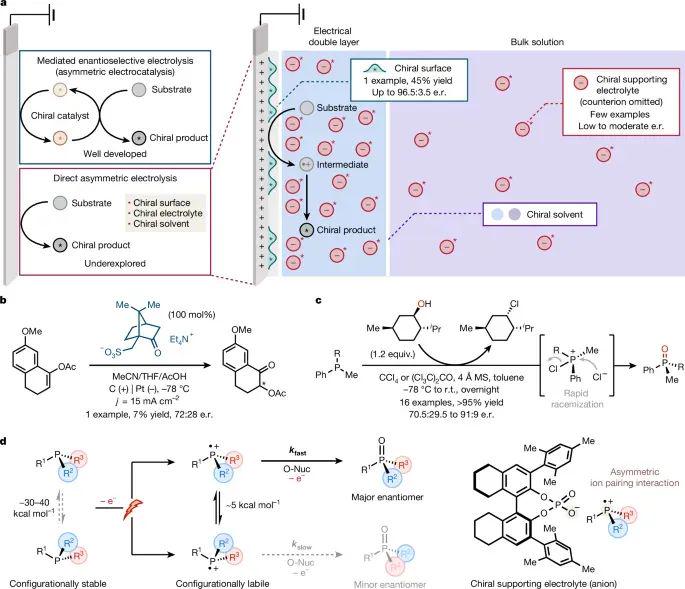
图 1. 通过手性支持电解质和三价膦的 DKR,通过膦基自由基阳离子的金字塔反转实现不对称直接电解
【有“导向基”底物的优化与范围】
研究团队以含酰胺吊环的模型膦 1a 验证方法。24 种部分饱和双萘基磷酸盐中,双间甲基取代的 CE3 在 40 mol% 用量时将产物 2a 的e.r.由60:40提升至70:30。随后筛选亲核试剂时,两类苯甲酰胺 Nu₂/Nu₃ 将e.r.进一步推高至93:7与95:5;二氯甲烷低介电常数的特性又最大化离子对势能井。在最佳条件下,多种底物(2b–2m)均以良好收率(45–76 %)获得91:9–95:5 e.r.,当通过重结晶纯化时可升至 99:1。值得一提的是68配资,产物 2m 经三步一锅还原可立体专一地回收近乎光学纯净的膦 (R)-1m,为后续有机催化或配体设计提供直接前体。
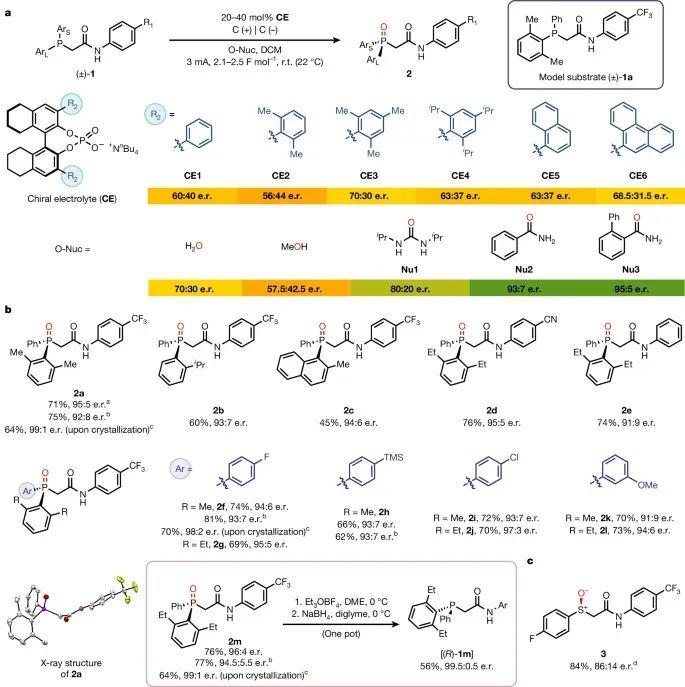
图 2. 具有导向基团的底物的优化和范围
【无“导向基”底物及流动电解扩展】
去除酰胺后,简单乙基取代的底物 4a 在水做亲核试剂时仅得71:29 e.r.。作者发现 o苯基苄醇 Nu₆ 凭借本身电化学还原副反应生成的甲苄烷副产物,可作为理想的O核,从而将 5a 的e.r.推至94:6并保持98 % 收率。改变膦上芳基或烷基后(5b–5h),多数产物仍维持86–94 % e.r.;环型膦(5i, 5j)稍降至≈90:10;三芳基膦 5k 则受位阻与缺乏αH影响,仅得71:29。晶化同样可将5a与5c提高到>99:1,并通过BH₃俘获专一还原为光纯膦 6。更具工业意义的是,作者在127 µm电极间距的连续流装置中,将CE3 负荷下探至20 mol%仍保持93:7 e.r.,显示出工艺放大与经济性的双重可行性。
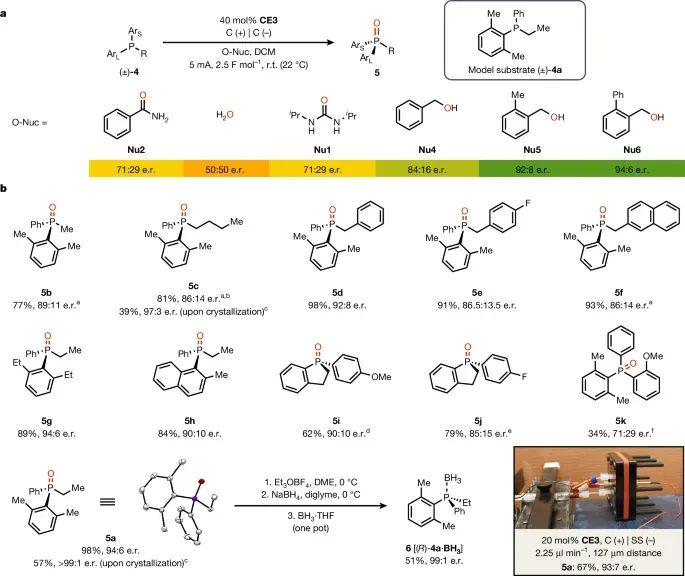
图 3. 不含导向基团的底物的优化和范围
【机理与计算能谱】
机理研究表明,膦首先在≈0.78 V(vs Fc/Fc⁺)被阳极单电子氧化为IN1,随后与亲核剂同步发生质子转移成键的协同过渡态TS2(ΔG‡ = 7.9 kcal mol⁻¹),其能垒高于磷鎓自由基的翻转(4.1 kcal mol⁻¹),因此符合DKR框架;随后的二次氧化与脱离均势能自由。相比之下,由开放壳层中间体引发的β 裂解需10.9 kcal mol⁻¹(TS3),故难以竞争。整体能谱描绘出“快速消旋慢速手性控制”的序列,印证亲核加成才是对映选择决定步骤。
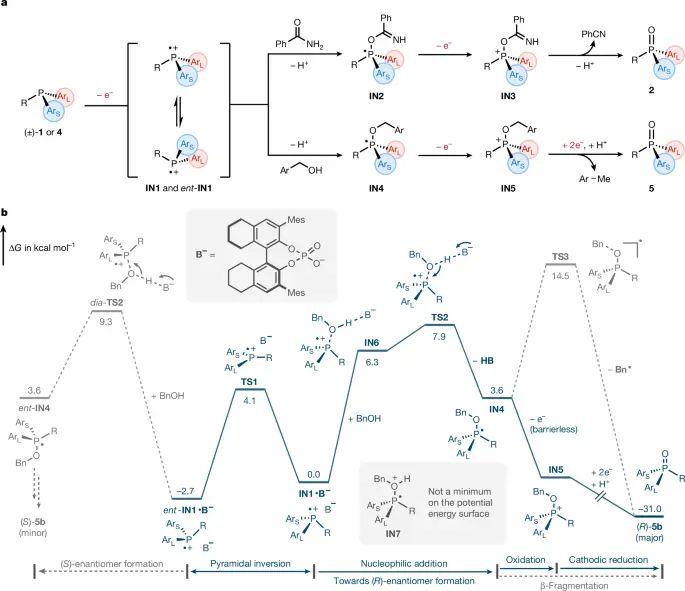
图 4. 提出的机制和计算能量图
【动力学与非共价互作阐释】
在1:1 混合的电子富集 Nu₇(4OMe 苄醇)与电子贫乏 Nu₈(4CN 苄醇)竞争实验中,产物 5a 仍给出89:11 e.r.,几乎复制Nu₇单独反应的87:13,说明亲核加成确为速率限定环节。时间分辨监测发现底物全程保持50:50 消旋,而产物e.r.随电荷通过量快速稳定,进一步佐证DKR。本构建中,一旦引入25 mol% 1,1二乙酰铁作为氧化媒介,氧化位置从界面转移至溶液体相,使2a的e.r.由93:7骤降至82:18;将CE3总量增至120 mol%后部分恢复到89:11,凸显电双层中手性离子富集的关键性。密度泛函展示的TS2与diaTS2仅差1.4 kcal mol⁻¹,却因TS2存在2.17 Å的C(sp³)–H···O氢键及CH···π、边面π堆积等多重非共价作用,而使diaTS2中对应距离拉长至2.33 Å,最终导致(R)产物优势输出。
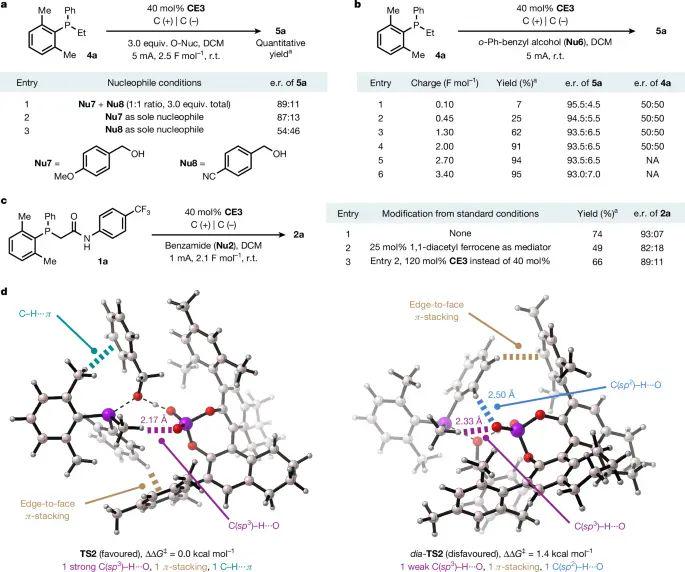
图 5. 手性支持电解质介导的电化学膦氧化机理研究
【总结】
本工作首度证明“手性支撑电解质+动态动力学拆分”可在无需外加手性催化剂的直接电解反应中实现>99 : 1的对映纯度。其可回收、亚化学计量的手性磷酸盐在流动电解中还能进一步节省用量,为构筑P‑手性膦氧化物及其还原后膦配体提供了一条高效、环境友好的新途径。作者预期,该策略有望推广至更多经离子‑自由基中间体的单电子转移反应,开启手性电化学合成的新篇章。
来源:高分子科学前沿
声明:仅代表作者个人观点,作者水平有限,如有不科学之处,请在下方留言指正!
鸿满仓配资提示:文章来自网络,不代表本站观点。
相关文章



沪深京指数
热点资讯